锂硫化学中的硫还原反应(SRR),即从S8冠状分子完全转化成Li2S,是一个16电子转换过程,其中涉及到一系列复杂的可变链长的多硫化物的转化。这种16电子的SRR过程对于高密度能量存储非常有意义,但是反应过程受到缓慢的硫还原动力学和多硫化物穿梭效应的困扰。研究人员已经在物理/化学吸附多硫化物等方面付出了相当大的努力来解决多硫化物的穿梭效应,但是这些被动限制策略从根本上不能完全防止多硫化物溶解到电解质中。虽然利用电催化来加速可溶性多硫化物中间体转化为不溶性Li2S2/Li2S已经被其他文章提出来是解决多硫化物穿梭效应的可行方法,但是硫还原反应的动力学还没有被深入探究,并且使用这种电催化效应来解决多硫化物穿梭问题的潜机理还没有被完全地阐释清楚。
研究目标:
1 硫还原反应的各步转化的反应活化能的建立;
2 催化硫还原反应的机理,动力学,反应活化能的本征研究;
3 密度泛函计算对催化硫还原的新理解;
4 提高锂硫电池的总体性能
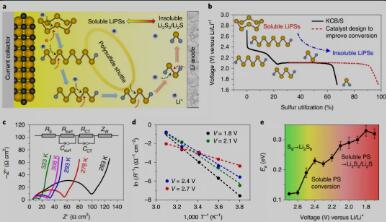
Fig. 1. Activation energy in sulfur reduction and PS conversion reaction.
多硫化物演化中的活化能势垒
硫还原反应过程涉及一系列多硫化锂(LiPSs)之间的多电子多步演化。S8冠状分子首先与锂离子2.7-2.4V的电压范围内形成长链Li2S8,随后通过S-S键的连续裂解,接着在2.3-2.1V范围内形成Li2S6或Li2S4,最后在~2.1-1.7V下转变为不溶性Li2S2和Li2S(Fig. 1a and b)。每一步的SRR动力学参数可以用活化能(Ea)来表示。为此,通过在不同温度下获取同一电压下的电荷转移电阻,我们得到转化过程中每一步的Ea(图1c-e)。结果表明,S8环分子的初始裂解被认为是一个相对容易的过程,而随后裂解成较短链的多硫分子变得越来越困难,转化成不溶性产物的最后步骤特别慢。由于大多数长链多硫化物可溶于电解质,并且这些多硫化物转化成短链的过程具有极高的活化能,从而导致了多硫分子在电解质中的积累、造成穿梭效应和快速容量衰减。为此,设计合适的电催化剂来降低这种能量势垒,并加速可溶性中间产物向不溶性Li2S2/Li2S的转化,将能有效解决穿梭问题。
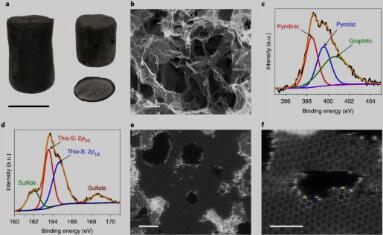
Fig. 2. Material characterizations of the N,S-HGF.
杂原子掺杂HGF催化剂的合理设计
我们选择了一系列杂原子掺杂的多孔石墨烯骨架(HGFs)作为模型催化剂来探索电催化SRR(Fig. 2a and b)。合成的多孔石墨烯结构既可以提供高效电子传递和离子传输的通道,也为杂原子掺杂提供了丰富的边缘位置。XPS表明氮和硫在石墨烯中的成功掺杂(Fig. 2c and d)。杂原子掺杂HGF中的键合结构也可以通过环形暗场扫描透射电子显微镜(ADF-STEM)直接验证。硫原子(图2e中的亮点)仅在纳米孔的边缘位点(~ 1-2nm)与碳原子以噻吩型键合的形式结合(图2f)。
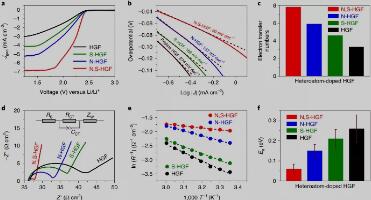
Fig. 3. Catalytic SRR activity and kinetic analyses of heteroatom-doped HGFs in RDE.
电催化SRR的活性、动力学和机理
图3a显示了不同杂原子掺杂的HGF样品的硫还原反应极化曲线,表明N,S-HGF的总体过电位较低。使用LSV曲线确定塔菲尔斜率(η)和交换电流密度(J0),N,S-HGF具有最小的η和最高的J0,表明更快反应动力学和更高的催化活性。为了理解催化剂存在下的硫还原机制,我们计算了反应过程中的电子转移数。N,S-HGF催化剂的表观电子转移数约为7.8,表明整个还原过程为8电子。相比之下,N-HGF、S-HGF和未掺杂HGF的电子转移数可分别计算为~5.9、~4.6和~3.3(图3c)。这些结果表明,NS-HGF可以促进更完全有效的硫还原和更快速地将多硫化物转化为不溶性产物。 电化学阻抗谱曲线(图3d)显示,与N-HGF、S-HGF和原始HGF相比,N,S-HGF催化剂在硫还原反应期间显示出最小的电荷转移电阻,表明其优越的电荷转移动力学。 根据阿伦尼乌斯关系,测量不同温度下的电荷转移电阻可以确定活化能。相较于其他的催化剂的活化能(N-HGF为0.09 eV,S-HGF为0.15 eV,未掺杂HGF为0.23 eV),N,S-HGF具有最低的活化能(0.06 eV) (图3f)。
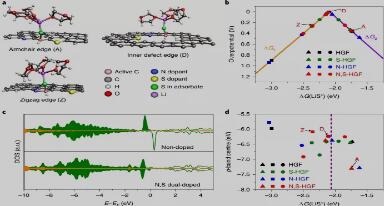
Fig. 4. DFT calculations on the activity origin of the heteroatom-doped HGFs on SRR.
电催化SRR的活性和动力学的理论理解
DFT计算表明,LiS自由基中间体在基面碳原子上的吸附能太弱,而在杂原子上的吸附能太强。因此,与杂原子相邻的碳原子提供了最佳的吸附位点,并且是催化硫还原反应过程最可能的活性位点。图4 显示了杂原子掺杂HGF活性的DFT计算结果。由于最终反应(Li2S2+2Li+2e-→2Li2S)是速率决定步骤,其Ea比其它转化步骤大得多,在研究不同可能结构的催化性质时,我们将计算重点放在最终的双电子过程上。首先,我们构建了更接近实际条件的微溶剂化模型(图4a)。 图4b的volcano图将最后一步的过电势与LiS自由基中间体在不同活性位点上的吸附能联系起来。结果表明,氮、硫双掺杂提供了更精细的调控,将NS-HGF体系几乎推到volcano图的顶部,并进一步将过电势降低到可忽略的值。 为了揭示高催化硫化还原反应活性的根源,我们认为掺杂过程可以精细调控催化位点的p轨道,从而实现催化性能(图4c)。基于此,我们使用p带中心作为活性炭上的状态密度,以描述掺杂杂原子的催化剂的电子结构,并发现了与LiS吸附能的关系(图4d)。
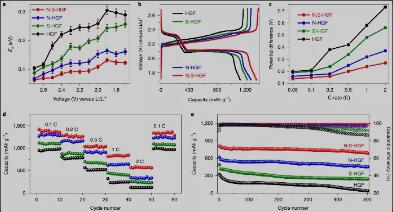
Fig. 5. Activation energy profiles and overall performance of the heteroatom-doped HGF cathodes in Li–S coin cells.
锂硫电池中的SRR
图5a显示了基于四种不同催化剂的电极在不同电压下的Ea曲线。随着杂原子掺杂剂的引入,活化能大大降低,特别是最终速率决定步骤。四种杂原子掺杂的HGF的不同Ea值也可以解释不同的极化电压间隙(图5b,c)。为了直接评估电催化剂对电池性能的影响,我们进一步比较了不同催化剂组装的锂硫电池电池的速率能力和循环行为。在硫质量负载为4mg cm-2的情况下,N,S-HGF电极表现出优异的速率能力,在0.1C、1C和2C条件下的比容量分别为1,390、840和577 mAh g-1(图5d)。此外, N,S-HGF电极也表现出优异的循环稳定性,在1C下500次循环时显示出极低的容量衰减(每圈0.025%),而N-HGF、S-HGF和未掺杂HGF的容量衰减分别为每圈0.054%、0.098%和0.162%(图5e)。这些特性都凸显了N,S-HGF的高效SRR催化活性对电池性能带来的极大提升。


 京公网安备
京公网安备